

So wird MPS Typ I vererbt
MPS I wird von den Eltern an die Kinder vererbt. Jedes Kind erbt von Mutter und Vater jeweils ein Gen, welches für die Produktion des Enzyms α-L-Iduronidase verantwortlich ist. Überträgt nur ein Elternteil den Gendefekt, bricht die Krankheit nicht aus. Das defekte Gen wird daher über einen „rezessiven Erbgang“ weiter gegeben.
Das Gen, das für die Produktion von α-L-Iduronidase zuständig ist, liegt nicht auf einem Geschlechtschromosom, sondern auf einem geschlechtsunabhängigen Chromosom, das in der Medizin als „Autosom“ bezeichnet wird.
Beim Vererbungsweg von MPS I sprechen die Ärzte daher von einem autosomal rezessiven Erbgang.
Bei Personen mit nur einem defekten Gen reicht das gesunde Gen aus, um genügend α-L-Iduronidase zu produzieren, so dass die Anreicherung des Speicherstoffs Glykosaminoglykan verhindert wird und die Krankheit somit nicht ausbricht. Diese Personen werden „Überträger“ genannt.
Haben Mutter und Vater jeweils ein gesundes und ein defektes Gen für α-L-Iduronidase, sind sie symptomlose Überträger, die entweder das gesunde oder das defekte Gen vererben können.
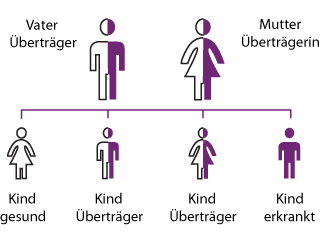
Bei jedem Kind besteht daher eine Wahrscheinlichkeit von 25 Prozent, die beiden defekten Gene zu erben und somit an MPS I zu erkranken.
Mit ebenfalls 25 Prozent Wahrscheinlichkeit erbt jedes Kind zwei gesunde Gene. Diese Kinder sind dann auch nicht Überträger der Krankheit.
Mit 50 Prozent Wahrscheinlichkeit werden jeweils ein defektes und ein gesundes Gen vererbt. Dieses Kind erkrankt selbst nicht an MPS I, ist jedoch ein „Überträger“.
Innerhalb einer Familie weisen alle erkrankten Geschwister den gleichen Enzymdefekt auf. Dennoch wurden sogar bei Geschwistern mit der gleichen Mutation verblüffend unterschiedliche klinische Manifestationen der Krankheit berichtet.
Die Wahrscheinlichkeit, ein defektes Gen zu haben, das MPS I auslösen könnte, ist sehr gering. Noch unwahrscheinlicher ist es, einen Partner zu haben, der den gleichen Gendefekt trägt. Das erklärt, warum MPS I eine so seltene und wenig bekannte Krankheit ist.